Medical Information
 |
|
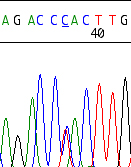
Example of an LDL receptor defect due to an known hete-rozygous G-to-A mutation, leading to stop codon at ami-no acid position 23: W023X, also called FH Cincinnati-5. Confirmation of the mutation by sequencing both strands. The LDL receptor activity is dramatically reduced to 2-5% of normal. Source: Original sequence from a patient examined at the diagene laboratories inc., Reinach, Switzerland. |
Familial Hypercholesterolemia
Familial hypercholesterolemia (FHC) is a relatively common genetic disease (prevalence 0.2% in the general population) leading to two to three times elevated cholesterol concentrations. High cholesterol concentrations within the blood cause typically changes in the blood vessels (atherosclerosis) eventually leading to heart attacks or cerebrovascular strokes. In familial hypercholesterlemia, these events occur typically before the age of 55 years in males and before the age of 65 years in females. Familial hypercholesterolemia is due to functional sequence variations in the gene encoding the low density lipoprotein (LDL) receptor. This protein is clearing the blood from cholesterol-rich low density lipoproteins. When these LDL particles remain in the blood for a longer while, they cause atherosclerotic lesions. An early diagnosis by the help of a gene tests and the early beginning of the treatment in affected individuals can prevent them from heart attacks and strokes. Since the patients have no symptoms from their elevated cholesterol concentrations, identification of family members from families with known familial forms of hypercholesterolemia is crucial. Thus, when identifying a patient with familial hypercholesterolemia, a family pedigree should be drawn and those individuals who might be affected should tested in the LDL receptor gene for mutations causing familial hypercholesterolemia. This concept of family tracing is an example for the highly successful strategies of protective medicine (see also PMF, Protective Medicine Foundation). So far, several hundred functional sequence variations have been detected worldwide. Therefore, in a new patient with the clinical features of familial hypercholesterolemia it is necessary to analyze the entire LDL receptor gene. Several laboratories offer the testing of a few more common mutations. However, testing for familial hypercholesterolemia makes it necessary to sequence the entire gene to detect also unknown functional sequence variations. In addition, it is important to compare the mutations detected with a comprehensive database that contains hundred known mutations that have been proven to be functionally important Thus, interpretation of such genetic results requires an extensive expertise. Testing of the LDL receptor gene, subsequent comparison with a proprietary database, and interpretation of the functionality of mutations discovered is available at diagene laboratories.
 |
|
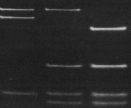
Example of a patient positive for the R3'500Q mutation in the apo B-100 gene. Source: Original sequence from a patient examined at the diagene laboratories inc., Reinach, Switzerland |
Familial defective Apolipoprotein B-100
Familial defective apolipoprotein B-100 (FDB) is the most prevalent monogenic lipoprotein disorder in Central Europe (prevalence 0.5% in Switzerland in the general population). Familial defective apolipoprotein B-100 is caused by mutations in the gene encoding the apolipoprotein (apo) B-100 molecule. Apo B-100 is part of the LDL particle and mediates as ligand the uptake of the cholesterol-rich LDL particle into the cell. LDL receptor defects as well as apo B-100 defects lead to a reduced uptake of LDL particles into the cell and thus, to hypercholesterolemia. Similar to familial hypercholesterolemia due to LDL receptor defects, cholesterol is elevated within the blood in patients with FDB. High cholesterol concentrations cause atherosclerotic changes and, as a consequence, heart attacks or cerebrovascular strokes. The most frequent mutation leading to familial defective apo B-100 is an arginin (R) to glutamin (Q) mutation at amino acid position 3'500 (R3'500Q) of the protein. A few further sequence variations in the gene encoding apo B-100 were discovered. As the testing for the most common mutations is simple, early diagnosis by the help of gene tests and early beginning of the treatment in affected individuals is easily possible and can prevent affected individuals from heart attacks and strokes. High cholesterol concentrations may develop relativey late in life (after the age of fourty years). Nevertheless, the risk to develop heart attacks and cerebrovascular strokes is similar to that in familial hypercholesterolemia. Hence, identification of affected family members from families with known familial defective apo B-100 is mandatory (see family tracing) and a major goal of protective medicine. In a patient with the clinical features of inherited forms of hypercholesterolemia, it is therefore very important to analyze the gene encoding apo B-100, the gene encoding the apo E (for exclusion/detection of familial dysbetalipoproteinemia, see below), and in a next stepthe gene encoding the LDL receptor (for exclusion/detection of familial hypercholesterolemia).
 |
|
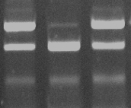
First lane: example of a patient positive (homozygous) for the R158C mutation (=apo E2/E2) in the apo E gene. Second lane: a patient positive having nor a mutation at amino acid position 112 nor at 158 (=apo E3/E3). Third lane: a patient positive (homozygous) for the C112R mutation (=apo E4/E4) in the apo E gene. Source: Original sequence from a patient examined at the diagene laboratories inc., Reinach, Switzerland. |
Familial Dysbetalipoproteinemia
Familial dysbetalipoproteinemia is caused by mutations in the gene encoding the apolipoprotein (apo) E molecule. Apo E is part of the intermediate density lipoprotein (IDL) particle and mediates as ligand the uptake of the cholesterol-and triglyceride-rich IDL particle into the cell. Homozygosity for the arginin (R) to cystein (C) mutation at amino acid position 158 (R158C) but also other, rare mutations lead to a reduced up-take of IDL particles into the cell and thus, to a combined elevation of cholesterol and triglycerides in the blood. Hence, similar to familial hypercholesterolemia due to LDL receptor defects and familial defective apo B-100 due to apo B defects, cholesterol concentations but also triglyceride concentrations are elevated in the blood. Combined cholesterol and triglyceride concentrations cause atherosclerosis and as a consequence heart attacks and cerrebrovascular strokes. The most frequent mutation leading to familial dysbetalipoproteinemia is the mutation at amino acid position 158 (R158C) on both alleles of the gene (also called apo E2 allele). Thus, familial dysbetalipoproteinemia is usually inherited in an autosomal-recessive fashion. Several further sequence variations in the gene encoding the apolipoprotein E were discovered and are associated with lipoprotein disorders. Genetic testing for these mutations is simple. Thus, early diagnosis by the help of a gene tests and early beginning of the treatment in affected individuals is easily possible and preserves affected individuals from getting heart attacks and strokes. Identification of family members from families with known familial forms of hyperlipidemia is recommended, although this disease is autosomal-recessively inherited although the chance to discover an individual affected by family tracing is lower than in dominantly inherited genetic disorders. In a patient with the clinical features of isolated hypercholesterolemia or a combined elevation of cholesterol and triglycerides, it is important to analyze the gene encoding apo E.
 |
|
Electrocardiogram of a patient with long-QT syndrome (a prolonged QT interval 570 msec). |
Long-QT syndrome
The Long QT syndrome is a hereditary disorder of the electrical rhythm system of the heart. The patients with long-QT syndrome have no symptoms until an often severe arrhythmia occurs that can lead to death. Hence, identification of members from families with known hereditary arrhythmia syndromes such as long-QT and Brugada syndrome is crucial (family tracing). This concept is also another example how gene tests and early treatment can prevent severe complications of a inherited disease (protective medicine). Long-QT usually affects children or young adults.
The electrical signal that stimulates the contraction of the heart muscle can be recorded on an electrocardiogram (ECG). A characteristic wave can be deduced; the different parts of this waveform are designated by letters (P, Q, R, S, and T). The interval between Q and T represents the time for electrical activation and inactivation of the ventricles. Patients with long-QT syndrome are susceptible to an abnormally rapid heart rhythm (arrhythmia) which can dramatically disturb the normal function of the heart and lead to death. A specific, very dangerous ECG pattern called "torsade des pointes" can then occur. The early beginning of the treatment, for example by using beta-blockers, in specific cases surgical procedures or implantable defibrillators, prevents patients from sudden death.
The gene test comprises full analysis of five genes
Since there are often large difficulties to identify the long-QT syndrome solely from measurement of the QT-interval duration on 12-lead ECGs, the role of gene tests in this disease is important. Scientific studies investigating the phenotype of patients with long-QT syndrome show that a normal QT interval (even when corrected for the heart rhythm, QTc) does not exclude this disease. Clinical diagnosis with measurement of QTc may be uncertain in as many as 50% of family members. In a substantial proportion of relatives from Long-QT syndrome families, molecular diagnosis is the most informative method. however, due to the enormous heterogeneity of the molecular basis of this disease, comprehensive screening for long-QT syndrome is difficult and can only be performed in specialized laboratories.